


Introduction
Agenus Inc. (NASDAQ:AGEN) is a small biotech pre-clinical company currently focused on developing cancer treatments. Founded by the current chairman and CEO, Garo Armen (PhD), in 1994, the company went public in February 2000 under the ticker AGEN. In the early 2000s, Dr. Armen gained significant popularity attention for his leadership at Elan Pharmaceuticals, a company he successfully steered away from the path of bankruptcy. He stepped down in 2005 amidst the withdrawal of a key drug from the market and significant investor frustration over the tanking stock price. Years later, following pipeline and legal turmoil, the company merged with Perrigo Company PLC, which remains listed on the exchange to this day.
Initially, Agenus primarily focused on anti-cancer vaccines, which often mirrored the common lack of efficacy associated with this therapeutic modality (Terry 2017) (Fan et al. 2023). However, Agenus’ immuno-oncologic approach prevailed. In January 2014, the company acquired 4-Antibody, a small biotech firm developing a platform capable of designing novel therapeutic antibodies with an existing portfolio of checkpoint modulators. Following the acquisition, Agenus developed a range of novel antibodies designed to treat cancer and quickly entered licensing deals worth hundreds of millions of dollars with companies such as Merck, Incyte, and Gilead. The company was developing checkpoint modulators (GITR, OX40, CTLA-4, LAG-3, TIM-3, and PD-1) through these joint ventures or independently. Despite this rich and collaborative pipeline, the company lost almost all its value during that period. In the last five years alone, Agenus’ stock has dropped by 70%, and an astounding 99% since its IPO. Earlier this year, the company underwent a 1-20 reverse stock split to remain compliant with NASDAQ listing requirements and to continue being eligible for inclusion in Russell indices.
Currently, aside from several licensing deals with low royalties paid to Agenus, the company is focusing on two key assets in its pipeline: Botensilimab (AGEN1181; anti-CTLA-4) and Balstilimab (AGEN2034; anti-PD-1). The former is particularly notable due to its consistent branding as “novel” and “next-generation Fc-enhanced.” We believe that careful consideration of these two primary assets is crucial for assessing Agenus as an investment, as their success or failure is likely to be the major determinant of the stock’s performance.
Update Note
This article was drafted prior to the press release outlining the FDA’s negative decision regarding the Bot+Bal BLA submission and the topline data of the phase 2 mCRC study. We anticipated the press release to surface towards the end of July; hence, while we chose to maintain the narrative of the article, several parts had to be updated based on the outcome of the meeting. Additionally, we provide an update paragraph, discussing the topline data. Readers should note that the FDA did not advise Agenus to file a BLA for AA in mCRC, a decision we argued would likely drive the stock price down in the short term. Following the press release, the stock opened down 60%, resulting in a market capitalization of approximately $150M (~cash basis).
Investment Thesis
We believe that Agenus is a company with assets that are not “next generation” enough to deliver breakthrough results and gain approval from regulatory agencies. To support our thesis, we will examine both pre-clinical and clinical characteristics of Botensilimab (Bot), Balstilimab (Bal), and Zalifrelimab (Zal), demonstrating that they compare similarly to the approved immune checkpoint inhibitors (ICIs) and the standard of care (SOC).
In our view, the poor management that led to the current valuation persists and is evident in the current decisions regarding the choice of indications to pursue. Some cancer settings in which Agenus is conducting trials are known to be unresponsive to ICIs, and others already have approved SOC ICI therapies. The company’s rationale is that the superior design of their novel antibodies can outperform currently available drugs; however, we do not share this view.
In addition to a pipeline with slim odds of success, the company has consistently struggled with capital. The recent licensing funding from Ligand is another cash injection that will help the sponsor continue developments; however, it will likely only last for another couple of quarters. Without securing significant clinical or regulatory milestones soon, the company is likely to face more financing hurdles.
As we anticipated, Agenus faced disappointment regarding the outcome of the recent metastatic colorectal cancer (mCRC) meeting with the FDA, where they hoped to receive positive news on potential accelerated approval (AA) based on the phase 1b and phase 2 studies. This outcome has shaped the short-term stock performance of Agenus. Consequently, we believe that developments in other cold tumors and ICI-refractory tumors will not prove to be exceptional, and the stock performance will reflect that. Additionally, the market opportunity of the mCRC sub-population chosen by the sponsor is relatively modest, suggesting that prior to the decline, the market was largely pricing in the AA.
Infra, we present the relevant background information and analysis of the available data on Bot, Bal, Zal, and other ICIs. Through comparative analysis of Agenus drugs with similar treatments, we intend to show that their “novelty” is largely over-exaggerated. Furthermore, we will highlight flaws in the company’s logic regarding the choice of indications and rationale for patient stratification. Our analysis will emphasize mCRC, as it is the most relevant short-term catalyst, but we will also review other cancer types targeted. Lastly, we will discuss cash-burn considerations and valuation estimates, to conclude that in our view, Agenus Inc is a sell.
Pipeline
In the latest 10-K and corporate presentation, Agenus mentions the following assets in their pipeline: antibody candidate programs (PD-1, CTLA-4, CD137, ILT2, ILT4, TIM-3, LAG-3, TIGIT, CD96, EP4, Hedgehog, CD205), QS-21 adjuvant, and allogenic iNKT. Notably, the majority of these programs are licensed to other companies, and Agenus is only eligible for development milestone payments or low royalties. Botensilimab and Balstilimab are considered “lead assets” due to the company owning a significant majority of the rights to potential future sales. Zalifrelimab, an anti-CTLA4 antibody similar to Botensilimab, despite being mostly owned by Agenus (licensed to Betta Pharmaceuticals in Greater China), appears to be currently halted in clinical development.
The CD137 antibody developments are primarily led by Gilead, and Agenus is eligible to receive a $50M exercise fee and up to $520M in potential milestones. The bispecific TIGIT program is led by BMS, with Agenus claiming eligibility for up to $1.34B in future milestone payments and mid-teen royalties. ILT-4 agonist development is facilitated by Merck, and Agenus is eligible for up to $85M in milestone payments and worldwide royalties. In 2018, Agenus sold all their rights to future royalties on QS-21-based vaccines for $190M; hence, despite registering the revenue on the income statement due for accounting purposes, they do not receive any additional payments related to sales of that product by GSK.
The iNKT platform shared with Minx, which Agenus owns approximately 62%, is still in the early developmental stage. The most advanced iNKT product is Agent-797, which presented mediocre data in a basket of 3L+ solid tumors (ORR monotherapy/combination/both: 0% (n=28)/17% (n=6)/3% (n=34)).
Our analysis will primarily focus on programs fully (or majority) owned by Agenus, specifically Botensilimab and Balstilimab. This focus is for several reasons. We believe that the results of operations regarding Bot and Bal are currently the most crucial for the company’s short-term success due to upcoming regulatory and clinical readouts and will also impact the company’s long-term performance as they are investigated in several early phase trials. We will not cover most of the shared programs due to their marginal relevance compared to Bot/Bal, considering the ownership of the assets. Furthermore, the recent funding agreement with Ligand granted Agenus $75M (possibly an additional $25M), but the deal involved the sale of a percentage of any future royalties from the shared programs and single-digit royalties on Bot/Bal. Hence, being a fractional part of Agenus’ business, the shared programs have become even less impactful. Despite this, we acknowledge that they are not worthless and could influence stock performance based on positive or negative outcomes and milestones; nonetheless, we believe they fall outside the scope of this article. The QS-21 royalties were sold for an upfront payment; thus, we do not find any considerations regarding that platform relevant, and the iNKT’s most advanced product has largely not impressed the market, reflected in the decline of the stock price of the daughter company and little to no response to treatment in the basket trial.
While preclinical considerations provide initial insights, their clinical significance is often limited. What truly matters to patients, physicians, and the regulatory agencies are the hard, clinical endpoints observed in human studies, as preclinical models, such as mice or cell culture assays, operate under homogeneous and controlled conditions that poorly replicate human biology? Nevertheless, we will briefly consider these preclinical factors for context and to assess whether the rationale for their use in the cancer types chosen by Agenus was valid. The preclinical data we present is just a portion of the figures provided by the sponsor. Displaying all the data would be beyond the scope of this article and unnecessary. Therefore, we have chosen the most relevant figures to support the claims regarding the molecules’ efficacy and their purported “novel” functionality.
Balstilimab
The advertisement for Balstilimab (anti-PD-1) by Agenus is not as prominent as that of Botensilimab, making our thesis here potentially less contentious. Based on the available data, we believe that Bal is not superior to the currently leading anti-PD-1 antibodies, such as Pembrolizumab or Nivolumab. The design of the molecule is identical to Pembro and Nivo, being an IgG4 antibody with an S228P Fc-region mutation (resembling IgG1), introduced to minimize Fab-arm exchange, and the affinity of the antigen-binding region specific for the PD-1 receptor on immune cells (Scapin et al. 2015). The pharmacodynamic profile, described by a set of kinetic rate constants (ka, kd, KD), is similar among the three antibodies, and Nivo was determined to be in the same epitope binding bin as Balstilimab (Brown et al. 2020).
The company presented various pre-clinical findings of Bal; however, they rarely compared the available anti-PD-1s directly. A poster presented in 2018 at AACR described a novel pharmacodynamic biomarker assay designed to test for antibody response to therapy. The experiment compared blood samples from patients treated with the Ipi+Nivo combo and the Bal+Zal regimen, establishing that a similar absolute increase in the biomarker was observed in both groups. Furthermore, the authors noted that no changes in total immune cell populations were observed after combination therapy in both regimens, suggesting a similar systemic activity profile. Despite evidence clearly indicating similar activity between the Agenus PD-1 antibody and currently marketed drugs, a poster titled “Differentiated activity profile for the PD-1 Inhibitor Balstilimab” was presented in 2021 at ASCO, suggesting an added benefit of Bal over Pembro/Nivo. Exhausted T-cell assays performed on tumor samples from two patients reported comparable results in the percentage of tumor cells killed at the endpoint, with often overlapping confidence intervals, shown infra.
Patient 1 Agenus Poster Differentiated Activity Profile for the PD-1 Inhibitor Balstilimab Patient 2 Agenus Poster Differentiated Activity Profile for the PD-1 Inhibitor Balstilimab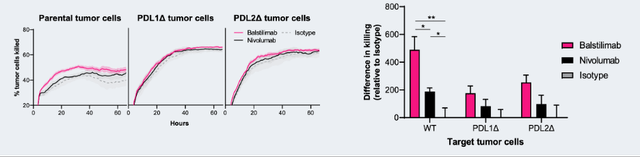
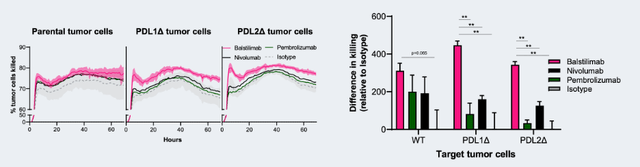
Notably, the significant p-values and higher pink bars on the right are presented as arguments for Bal’s superiority, but in our opinion, they do not hold significant meaning. In both samples, the percentage of cells killed at the last time point was comparable, which seems to be a much more relevant data point than the “area under the curve” represented by the bar plots on the right. Furthermore, it is unclear why sample 2 was tested with Pembrolizumab and sample 1 was not, which undermines the credibility of the presented data. Overall, we conclude that Bal does not differentiate itself meaningfully from the currently available anti-PD-1 antibodies, such as Nivolumab and Pembrolizumab.
Pre-clinical considerations are important when evaluating the rationale of the therapy, but they are merely surrogates for clinical findings. Balstilimab has been extensively investigated by the sponsor, and the closest the company got to filing for approval was for Bal monotherapy in metastatic cervical cancer (mCC).
In April 2021, Agenus filed a Biologic License Application (BLA) for accelerated approval of Bal in recurrent or metastatic cervical cancer with disease progression on or after chemotherapy, based on a pivotal phase 2 single-arm trial and preclinical data that, per the press release, “suggest that Balstilimab demonstrates differentiated features from other anti-PD-1 antibodies”. The study enrolled 161 patients, of which 140 received Bal treatment and were included in the efficacy analysis (O’Malley et al. 2021). In the cohort of 140 patients, the ORR was 15%, and when stratified for PD-L1 positive/negative status, the rates were 20% (n=85) and 8% (n=38), respectively. Notably, 17 patients had an unknown PD-L1 status, and only 1 person in that cohort (6%) had an objective response. Treatment-related adverse effects of grade 3+ occurred in 12% of patients, and adverse events leading to discontinuation of treatment occurred in 4.3%. Only four months later, Agenus, on the advice of the FDA, withdrew the submission due to the accelerated approval of Pembrolizumab in the same designation. The study on which Pembro was granted approval, KEYNOTE-158, enrolled and treated 98 patients with Pembro and reported an ORR of 12%, stratified by PD-L1 positive and negative status, with response rates of 15% (n=82) and 0% (n=15), respectively (Chung et al. 2019). Unlike the Bal trial, there was only 1 patient with unknown PD-L1 status. Treatment-related adverse effects of grade 3+ occurred in 12% of patients, and adverse events leading to discontinuation of treatment occurred in 4.1%.
Despite Agenus’ claims of superior activity, approval was granted to Pembro; perhaps the FDA did not share their view on differentiated activity of the two compounds. We view the minor differences in outcomes as arising due to chance and the small size of both trials, considering the similar dosing regimens, the same mechanism of action, and the higher rate of patients with unknown PD-L1 status in the Bal trial. Overall, we consider this sufficient evidence of the lack of significant difference between the clinical activity of Balstilimab and Pembrolizumab.
In addition to the monotherapy regimen, Agenus investigated a combination therapy of Balstilimab with Zalifrelimab (anti-CTLA-4) in mCC. The PD-1 plus CTLA-4 combination is known to yield stronger responses at the cost of more toxicity associated with greater stimulation of the immune system. The combined regimen reported an ORR of 26% in the cohort of 125 evaluable patients (O’Malley et al. 2022). The response rates, classified by PD-L1 status, were 33% for the positive group and 9% for the negative group. There was a significant number of patients with unknown PD-L1 status (n=25), who had an ORR of 28%. The increase in observed responses came at the cost of increased toxicity; treatment-related adverse effects of grade 3+ occurred in 20% of subjects, and adverse events leading to discontinuation of treatment occurred in 7.7%. Interestingly, in the discussion, the authors comment on the findings from the CheckMate 358 trial, which used the Ipi+Nivo regimen in mCC (Oaknin et al. 2024). In a similar cohort dosed using a comparable regimen, the ORR of Ipi+Nivo was 26% (n=27), with the high-dose Ipi group reporting an ORR of 30% (n=20). The progression free survival (PFS) figures cannot be reliably compared between trials as they include patients treated in the first line in the CheckMate 358 trial, as opposed to only second-line patients in the Bal+Zal study. We argue that these comparable results of Bal+Zal and Ipi+Nivo in mCC further suggest that these compounds are clinically similar.
In summary, we believe Balstilimab does not significantly, if at all, differ in functionality from Pembrolizumab or Nivolumab. Findings from Zalifrelimab in combination with Balstilimab also suggest that Zal does not exhibit any profound differentiated activity from other anti-CTLA-4 antibodies, which we discuss infra.
Zalifrelimab
Zalifrelimab is one of the two IgG1 anti-CTLA-4 antibodies owned by Agenus, which we will cover in this article. Currently, Zal is not the primary focus of the company’s pipeline, as developments have been temporarily halted according to the latest corporate presentation. We chose to carefully consider Zal data to support one of our major claims: Bot is, at best, slightly different from Zal and, at worst, nearly identical. Since Zal was halted and did not differentiate itself from other CTLA-4 antibodies, we believe Bot will follow the same trajectory. This is particularly important given how both drugs are described by the company. Zal is simply referred to as “CTLA-4,” while Botensilimab is called “Fc-enhanced CTLA-4,” “novel,” and “next-generation.”
Agenus Inc Publications Website
Knowing that Zal combined with Bal in mCC led to outcomes comparable to other CTLA-4 antibodies could lead one to assume that, if Bot and Zal have the same design, both share a similar clinical profile. The main differentiator, “Fc enhancement,” is neither novel nor unique to Bot, as both were reported to share the same “enhancement.”
The modification in question refers to a set of three mutations in the Fc region of the antibody, collectively referred to as DLE (S239D/A330L/I332E), designed to enhance binding between the region and Fcγ receptors found on immune cells. To fully appreciate the complexity and functionality of Fcγ receptors, a thorough understanding of immunology is beneficial. For an unfamiliar reader, it can be simply explained that when Fcγ receptors are bound, they modulate the activity of immune cells, either enhancing or diminishing their activity, depending on the specific receptor and co-binding molecules. While the Fab fragment is responsible for antibody specificity and targeted binding, the Fc region structure can impact immune effector functions (interaction with immune cells, complement binding) and the antibody’s half-life in plasma (Abdeldaim and Schindowski 2023). The first considerations of Fc structure-function relationships were researched in the early 2000s, and the DLE Fc subtype was initially mentioned in the literature in 2006 (Liu et al. 2020). Clearly, this “novel” “Fc-enhanced” antibody that Agenus has is not novel at all; in fact, the design principle is almost 20 years old. Furthermore, there are several different mutation combinations and sugar modifications that lead to varying Fc cellular effector functions.
Fc-region Antibody Engineering – doi.org/10.3390/antib9040064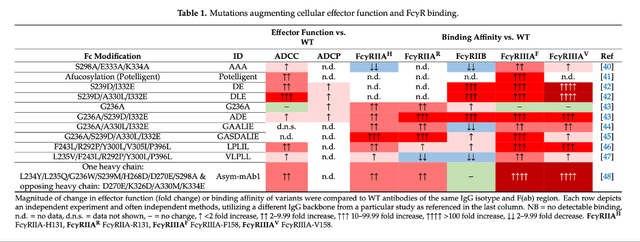
The rationale for DLE is the increased binding to FcγRIIIA, and as outlined in the figure supra, this results in increased antibody-dependent cellular cytotoxicity (ADCC), allowing for more efficient removal of cancerous cells by effector T-cells and, additionally, APC-mediated T-reg cell killing. What remains to be determined is whether these changes bear any clinical significance.
Notably, while DLE modification enhances ADCC, it also increases affinity for FcγRIIB, the only inhibitory Fcγ receptor found in humans, which could introduce unwanted effects. In contrast, sugar-modified antibodies have increased effector function without FcγRIIB binding ability.
It is important to note that these mutations are far from irrelevant; in many cases where the pathways are well-defined, the modifications can have a profound impact on outcomes. Several antibodies with Fc-enhanced regions have been approved over the years, but historically, there are examples of Fc-enhanced antibodies that clinically failed when compared to their “first generation” counterparts. Ocrelizumab, an antibody engineered for increased ADCC, failed to show improvement over the classic marketed equivalent, Rituximab (Liu et al. 2020). Another engineered drug, Margetuximab, despite increased FcγRIIIA and decreased FcγRIIB binding, showed a complete lack of improvement in overall survival (OS) in the breast cancer patient population (Rugo et al. 2022).
Our contention is that Zal, and by extension Bot, are not molecules with novel designs that represent groundbreaking advancements in the cancer space (similarly to Ocrelizumab and Margetuximab). Instead, they are marginal modifications described decades ago that may or may not impact human clinical outcomes. It is important not to confuse a modification leading to a clear change in functionality, such as a Fc mutation in a PD-1 antibody resulting in a lack of binding to a specific receptor, with a modification like DLE, which skews binding affinities for several likely interdependent receptors with pathways we often lack complete understanding of. What does the data provided by the sponsor suggest about Zal’s activity?
An article sponsored by Agenus shows that the modified antibody resulted in about 20% T-reg cell lysis, compared to about 5% in the isotype control (Gombos et al. 2018).
T-Reg cell lysis assay – doi.org/10.1371/journal.pone.0191926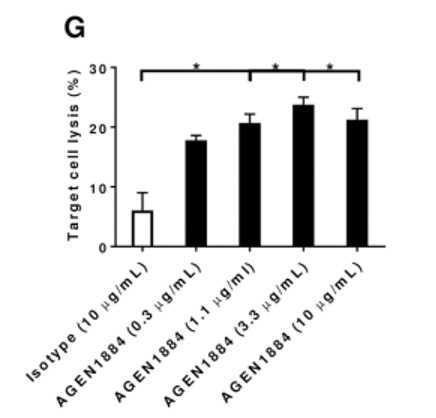
This figure fails to compare the effect to any other commercially available CTLA-4 antibody, which is highly relevant considering the claims of a differentiated profile from other CTLA-4 antibodies like Ipilimumab, which was also reported to deplete T-reg cell populations (Arce Vargas et al. 2018). Furthermore, there appears to be an almost immediate plateau of target cell lysis regardless of the concentration used in the assay. Most importantly, the 20% lysis observed under idealized assay conditions raises questions about whether such a reduction would have a clinically relevant effect in a real-world setting. In other words, assuming a patient experiences a reduction of T-reg population in the tumor environment, will that have a profound impact on the tumor mass? We argue it is highly unlikely.
Pre-clinical findings shared by Agenus suggest that Zalifrelimab shares similar pharmacokinetic properties to Ipilimumab and Tremelimumab (interstudy comparison). The ka, kd, and KD values reported by Agenus were comparable to literature-reported values for Ipilimumab and Tremelimumab (He et al. 2017). Given the pharmacokinetic profile, depleting properties, and unremarkable clinical findings, we find Zal to be, rather than a next-generation immunotherapy agent, at best a marginally improved anti-CTLA-4 antibody. Is Bot any different?
Botensilimab
Botensilimab is currently the key asset in the Agenus pipeline, accounting for the majority of owned programs, representing several near-term catalysts. We have already introduced Zal, and since we believe that Zal and Bot are highly similar, many of the considerations mentioned supra also apply to Bot.
Bot is an IgG1 anti-CTLA-4 antibody, with its Fc region engineered to bear DLE mutations. Since the antibody functions based on the same principles as Zal, the same rationale applies: stronger FcγR binding, T-reg depletion, and increased ADCC. There is little independent literature available on Bot; however, the company provides several pre-clinical figures showcasing the antibody’s functional characteristics. Most relevant are the T-reg-depleting findings, which, in the in-vivo mouse model, seem to exhibit a higher level of activity compared to previous findings discussed for Zal.
Fc-enhanced anti-CTLA-4 Antibody, AGEN1181: New Mechanistic Insights for Potent Antitumor Immunity and Combination Potential in Treatment-resistant Solid Tumors; T-Reg depletion Bot mouse In-vivo model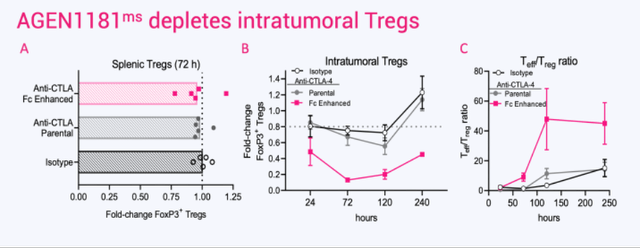
Nonetheless, the sponsor does not provide direct comparisons to Zal or other commercially available ICIs, making it difficult to assess if the assets differ significantly. The apparent greater reduction in T-regs might be due to interstudy comparisons, different protocols, or simply the different use of assays and in-vivo models. Considering that Zalifrelimab has the same mutation in the Fc region, one would expect similar activity regarding immune cell stimulation. In addition to animal model data, Agenus provides histologic slides from two cancer cases, thyroid cancer and sinonasal SCC.
AGEN1181, an Fc engineered anti-CTLA-4 antibody, demonstrates clinical activity, alone or in combination with balstilimab (anti-PD-1), and broadens the therapeutic potential of CTLA-4 therapy; Human tumor histology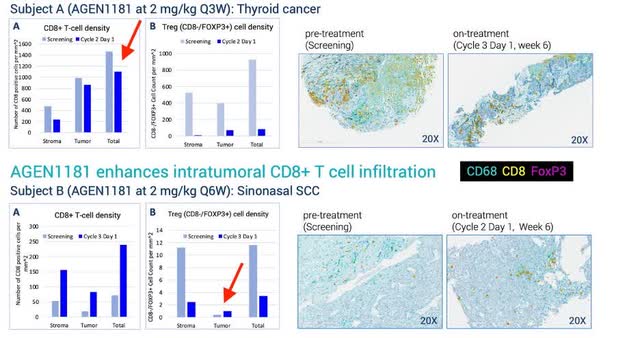
In subject A, Figure A shows a small decrease in cytotoxic CD8+ T-cells, which contrasts with the animal model findings of increased CD8+ proliferation post-Bot administration in mice. Subject A also experienced a notable decrease in the T-reg population, both in the stroma (outside the tumor mass) and within the tumor mass. Conversely, Subject B experienced a significant increase in the cytotoxic T-cell population and a depletion of the T-reg population. Notably, T-regs were largely decreased in the stroma, while intratumoral cells slightly increased over the 6-week period, suggesting mixed findings. This inconsistency, possibly due to the different cancer types, is nonetheless concerning. It indicates that the immune response to Bot may vary significantly between patients, raising questions about the predictability and reliability of the treatment’s efficacy across different tumor environments.
Overall, while both Bot and Zal exhibit several functional qualities that can impact the dynamics of the cancer immune microenvironment, including T-reg depletion, it remains questionable whether the animal model data and surrogate measures such as changes in effector cell ratios or immune cell proliferation will translate to meaningful clinical outcomes. As seen in the Bal+Zal mCC trial, the Fc-enhanced antibody seems to perform similarly to the commercially available PD-1 and CTLA-4 inhibitors.
Infra, we will discuss the findings of Bot and Bal efficacy from clinical trials in metastatic colorectal cancer, pancreatic cancer, and melanoma. We will also carefully examine and critique the recent publication of the phase 1b trial of Bot+Bal in mCRC and speculate on the possible outcomes of upcoming phase 2 trial data and the FDA meeting. While we intend to largely focus on mCRC, each cancer type will have a separate paragraph discussing the rationale for efficacy and contextualization to other ICIs.
Clinical Data
Most of Agenus’ clinical findings for the Bot, Bal, or Bot+Bal regimen in solid tumors are based on the C-800-01 basket trial. To date, and to the best of our knowledge, these antibodies have been administered to patients with ovarian cancer, sarcoma, melanoma, cervical cancer, pancreatic cancer, colorectal cancer, and lung cancer. Many of these cancer types are considered immunologically “cold,” meaning they are poorly responsive to immunotherapy due to their specific biology (Bonaventura et al. 2019). The lack of response to immunotherapy in these cold tumors is well-documented, with many trials of PD-1/CTLA-4 regimens using already approved antibodies having failed in the past (Ouyang et al. 2024). Agenus argues that they are focusing on these cancer types precisely because they are “cold,” asserting that Bot, being “Fc-enhanced” and a next-generation anti-CTLA-4 antibody, will show efficacy in patients who are normally not responsive to ICI. However, we do not believe there is sufficient pre-clinical rationale to justify this claim with high certainty, and there is currently a lack of clinical data to support these further investigations.
The sponsor also targets certain “hot” tumors, like melanoma and lung cancer, which are known to respond to ICI (Wang et al. 2023). Instead of demonstrating that their molecule can outperform the “old-generation” commercially available antibodies head-to-head, Agenus has chosen to investigate their ICIs in patients who are refractory to previous PD-1/CTLA-4 therapy.
Aside from Bal and Bal+Zal investigations in metastatic cervical cancer, which we discussed supra, clinical data on Agenus antibodies have been published recently published in nature medicine (excluding abstracts) (Bullock et al. 2024). This paper focuses on mCRC, which is also the closest program to reaching a regulatory meeting with the FDA, scheduled for July. Clinical data on other cancer designations have been published through press releases and journal abstracts. Therefore, we will devote most of our considerations to mCRC.
Colorectal Cancer
Colorectal cancer is a common neoplasm that affects parts of the colon or rectum (De Falco et al. 2019). Based on the location, the cancer is subdivided into right and left, with the two types showing differing biology, metastatic patterns, and responses to treatment (Adrià Cañellas-Socias et al. 2024). Early-stage disease can often be surgically removed, and there is a high chance that the cancer will not recur, effectively classifying the patient as cured. However, in approximately 50% of cases, the cancer will recur, and metastatic disease has poor prognostic statistics. The 5-year overall survival is about 60%, and the median OS in the metastatic setting is around 30 months (American Cancer Society 2023) (De Falco et al. 2019). Patients are treated with various chemotherapeutic agents, targeted therapeutics (VEGFR, EGFR), and most relevant to our considerations, ICIs.
Being generally considered a “cold” cancer, immunotherapy has historically shown little response in mCRC patients. However, a highly mutagenic subtype of mCRC cancers, known as high microsatellite instability (H-MSI) and deficient mismatch repair (dMMR) (about 5% of patients), has shown great responses to ICI treatment, resulting in classification as first-line (1L) treatment for patients with that cancer biology (Morris et al. 2022) (Cervantes et al. 2022). This response is commonly attributed to the high mutagenic load of the cancer cells, which present many “neoantigens” (new markers) on their surface, making them readily recognizable and destroyable by immune cells (also referred to as high TMB). The less-mutagenic subtypes, proficient mismatch repair and microsatellite stable (pMMR/MSS), have historically shown little to no response to ICI therapy (Ganesh 2021).
Agenus set out to test their “novel” antibodies in patients historically non-responsive to such treatment, owing to the alleged properties that could “warm” the cold mCRC. They chose to study the drug in a third line (3L) regimen, effectively competing with two therapies currently used in that setting, Regorafenib (Stivarga) and trifluridine-tipiracil (Lonsurf).
Why did they choose not to test their drug against Pembrolizumab, Ipilimumab, and Nivolumab in H-MSI/dMMR patients? We can only speculate, but we doubt that Bot and Bal would show improvement over the other commercially available ICIs. We also believe that the Bot+Bal regimen is unlikely to be superior to Ipi+Nivo in MSS/pMMR patients for several reasons. As previously mentioned, we are skeptical whether Fc-enhancement can induce an immune response in patients with tumor biology that is not responsive to ICIs. Besides T-reg depletion, other immunosuppressive mechanisms are not addressed by Bot’s scientific rationale (Adrià Cañellas-Socias et al. 2024). Cell types such as cancer-associated fibroblasts (CAFs) can secrete cytokines and extracellular matrix, which exhibit immunosuppressive activity. Additionally, the architecture of the tumor itself can render ICI therapy useless due to the exclusion of T-cells from the cancer site (immune deserts).
To date, Agenus has published the phase 1b data from the C-800-01 study and intends to present phase 2 study data to the FDA during the upcoming meeting, hoping to secure a BLA filing in anticipation of AA. We will first carefully consider the data from the published trial and later speculate on the data from the phase 2 study. Lastly, we will consider the possible outcomes of the FDA meeting.
The C-800-01 study was an open-label dose escalation trial designed to assess the efficacy of Bot/Bal in a basket of solid tumors. In the dose escalation part, 83 patients received Bot or Bal regimens, and 10 of those 83 patients were mCRC MSS. After the escalation part, the study enrolled an additional 396 subjects, with 143 MSS mCRC patients receiving at least one dose of the Bot+Bal regimen. In total, the authors report that 148 subjects, 6 from dose escalation and 142 from expansion cohorts, were mCRC MSS and treated with the Bot+Bal high-dose regimen. Out of these 148 patients, the authors chose to evaluate responses in only 101 who completed at least 6 months of follow-up.
Several interesting observations can be made based on the baseline characteristics of the evaluable patient cohort.
Baseline data – doi.org/10.1038/s41591-024-03083-7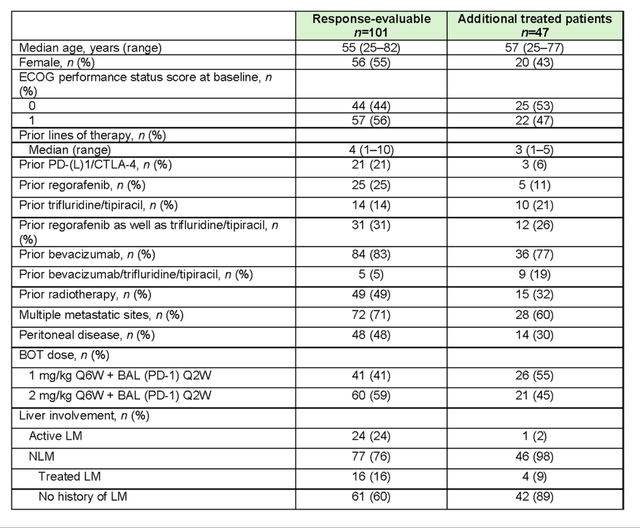
The patients were, on average, in their mid-50s, indicating a younger cohort. There was approximately a 50/50 split between ECOG scores of 0 and 1, with patients having a median number of four prior therapy regimens. Additionally, 50% of them had peritoneal metastasis. The NLM (non-liver metastatic) subjects made up about three-quarters of the evaluable cohort and almost the entire unevaluable cohort.
The sponsor chose to enroll only NLM patients after a certain time-point, noting better efficacy in this sub-population. NLM patients were further divided into those with treated liver metastasis (LM) and those with no history of LM. The decision to classify patients with treated LM as NLM is questionable. If patients without liver metastasis possess different cancer biology that responds to ICI, then patients who had liver metastasis that was surgically resected should have similar cancer biology to those with unresected liver metastasis. This stratification subtly divides patients into sicker and healthier groups-patients with resected liver metastasis are likely healthier than those too sick to undergo surgery. Similarly, patients with no history of LM might have different cancer biology (not necessarily implying ICI responsiveness), but it is also highly plausible that their cancer biology is less aggressive and more manageable, making them “healthier.”
Our independent analysis of SEER*Stat cancer database on survival outcomes in LM and NLM CRC patients who underwent at least one chemotherapy regimen shows that NLM patients survive almost twice as long compared to the LM cohort (~24 months vs. ~50 months; n=48,989).
SEER*Stat Version: 8.4.3 Registries, Nov 2023 Sub (1975-2021) – Linked To County Attributes – Time Dependent (1990-2022) Income/Rurality, 1969-2022 Counties, National Cancer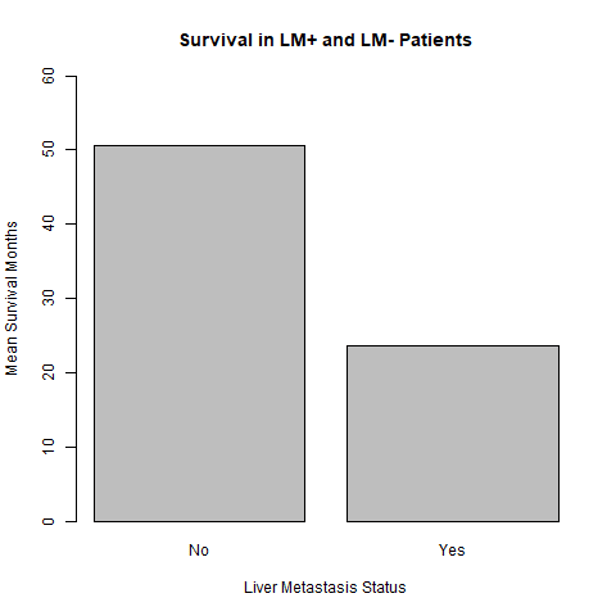
The study reports a gap in biomarker testing, which we consider a significant flaw for an ICI trial.
Biomarkers – doi.org/10.1038/s41591-024-03083-7
TMB, PD-1L cancer histology, and neoantigen load are all strong predictors of ICI efficacy (Ganesh 2021). The primary hypothesis behind ICI efficacy in dMMR and H-MSI mCRC is the neoantigen load associated with high TMB. Having biomarker data available for less than one-third of patients is a significant issue, especially considering that many FDA-approved regimens require biomarker testing for eligibility (Chang et al. 2021). This data allows for further stratification of patients more likely to respond to the treatment. However, in the phase 1b trial, Agenus does not provide sufficient data on these biomarkers.
The reported ORR in the evaluable cohort was 17%, with one complete response (CR) and 16 partial responses (PR). The median progression-free survival (mPFS) was 3.5 months, and the median overall survival was 20.9 months. The NLM cohort accounted for all the responses, with a rate of 22%, and reported a median PFS of 4.1 months and a median OS of 20.9 months.
Waterfall diagram ORR – doi.org/10.1038/s41591-024-03083-7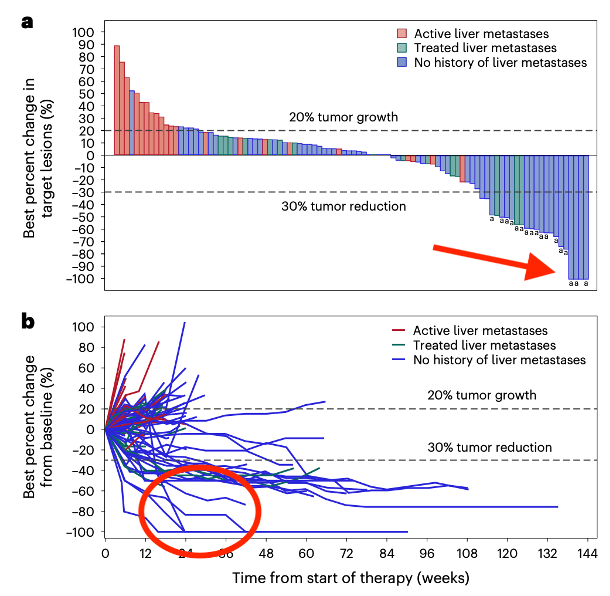
The findings in Figure A reveal a result that we consider unexplained in the text. The authors denote confirmed RECIST responses with the letter “a,” and five subjects that achieved a change of -30% or more are shown but not accounted for in the 17% ORR figure. While this is acceptable, it is peculiar that the diagram indicates 4 CRs; however, only 1 is reported in the text, despite 3 of those 4 being marked with the letter “a.” We speculate that these patients experienced a CR in clinical imaging, but later progressed or discontinued treatment. The same four patients are also shown in Figure B, with no signs of later progression on the graph. What happened to those patients, and why were they not considered CRs? This same discrepancy is replicated in the most recent corporate presentation, where despite showing 4 CRs on the diagram, the table next to the figure reports only 1 CR.
PFS – doi.org/10.1038/s41591-024-03083-7 OS – doi.org/10.1038/s41591-024-03083-7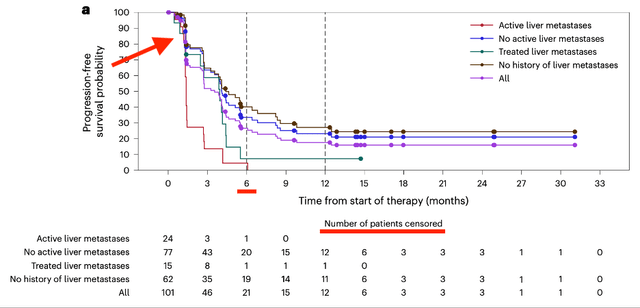
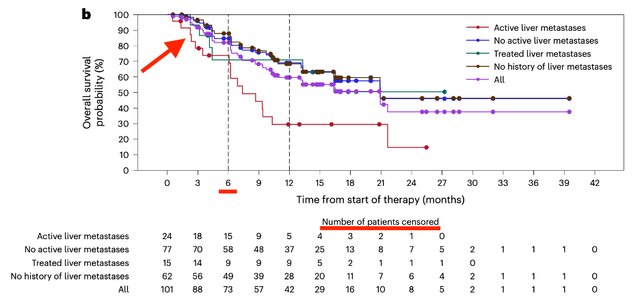
The PFS and OS Kaplan-Meier (KM) curves exhibit several questionable aspects that cast doubt on the results.
Both figures refer to “patients at risk” as “number of patients censored,” which are, strictly speaking, two different cohorts. Additionally, the PFS data show a significant amount of censoring at around 6 weeks, approximately when the first scan should be carried out to assess disease progression. Importantly, there is no censoring in the liver metastatic group; it is only the NLM cohort that collectively had about 10% of patients censored. The most plausible and concerning explanation for this observation is that the patients were experiencing adverse effects. The toxic regimen of combination immunotherapy could have made many patients unable to attend the scan; hence, they got censored. Assuming this is true, one of the two principles of KM curves would be considered violated-non-informative censoring. It is clear that patients unable to attend the first scan differ significantly from those who can, making it incorrect to assume the latter group would have the same outcomes as the former.
Early censoring is common with PFS figures due to the nature of the combined progression and survival endpoint. Overall survival KM curves are normally not prone to such censoring, as survival can be assessed even if the patient drops out or does not follow up. Surprisingly, in the OS data, there is significant censoring prior to month 6, despite the fact that the cohort analyzed was chosen based on having at least 6 months of follow-up. We believe that the patients censored in the first 6 months shown on the curve are likely those who were so sick that they discontinued the treatment and stopped subsequent follow-ups. Normally, these patients should not be censored, since investigators can still obtain the relevant OS information even if the patients discontinue the treatment. Here, we see this information was not accessed, which is worrisome.
The early censoring due to toxicity is also supported by the provided safety data. The supplementary figure reports that 55% of subjects had to interrupt treatment to deal with side effects, and 34% had to discontinue due to treatment-emergent adverse events.
Safety Data – doi.org/10.1038/s41591-024-03083-7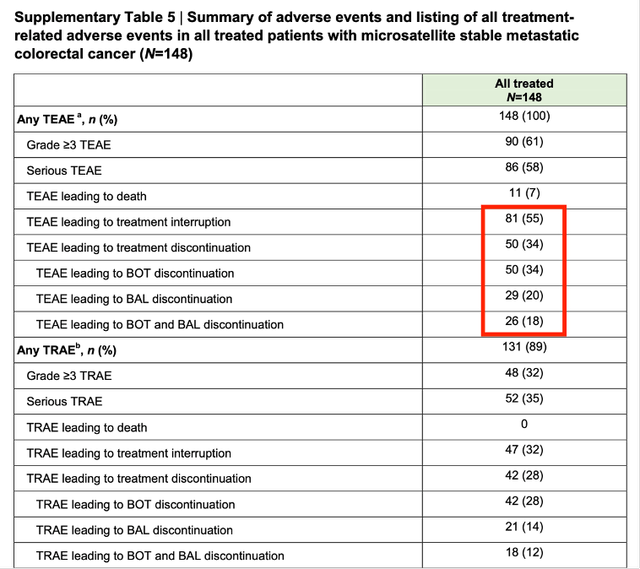
The toxic profile of Bot+Bal is typical of ICI combination therapy, characterized by a high rate of grade 3+ treatment-related adverse events (TRAE), including diarrhea, pyrexia, elevated liver enzymes, and intense inflammation of various organs. It is also worth noting that the total number of patients who discontinued treatment and the subgroup discontinuation figures do not add up, as illustrated infra.
Python – Venn diagram TEAE discontinuations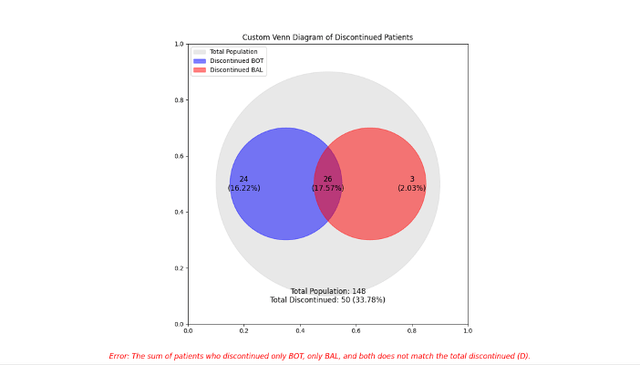
As shown below, the incidence of adverse events in the trial is notably high. Given the short follow-up period and the ongoing maturation of data, we expect that the frequency of these events will increase, especially for rarer occurrences that typically take longer to manifest.
TRAE list – doi.org/10.1038/s41591-024-03083-7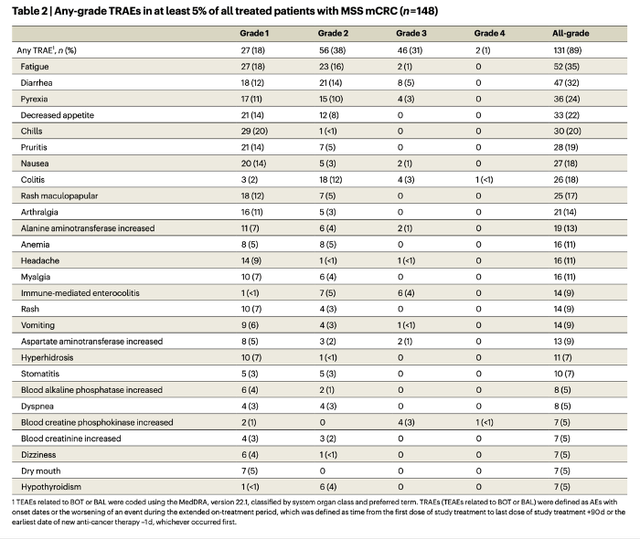
The data is still maturing, and it is possible that the authors, because of the quickly moving timeline, either did not attempt or were unable to obtain the relevant information about the censored patients. Regardless, the fact remains that the OS figure, which is by default exploratory due to the small size of the trial, is highly unreliable.
The elevated median OS (mOS) figures are likely influenced by informative censoring and the inherent quality of the NLM cohort. The data for the LM cohort aligns closely with findings from large studies investigating Regorafenib and trifluridine-tipiracil, leading us to believe that the observed improvements in OS in the phase 1b Bot+Bal study are not due to the drug’s efficacy, but rather the selection of a healthier cohort. A recent meta-analysis of outcomes in NLM patients supports this, showing that the PFS and OS can be comparable to those observed in the Bot+Bal trial.
PFS and OS in 3L mCRC patients – https://www.ejcancer.com/article/S0959-8049(24)00816-5/abstract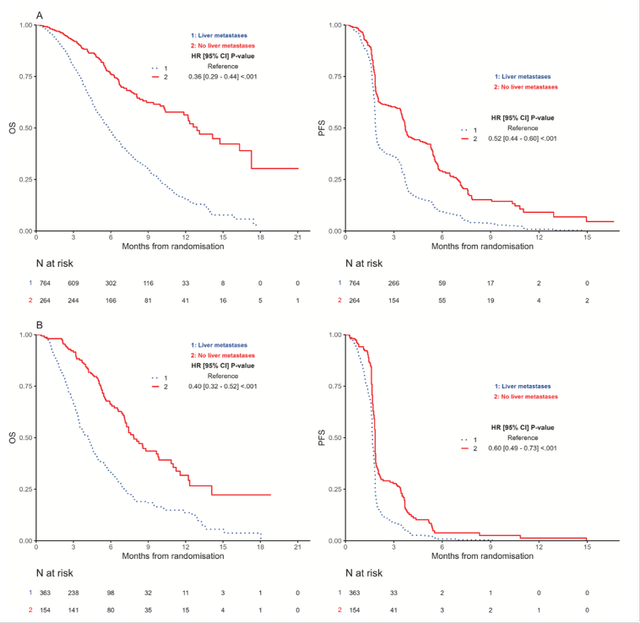
Clearly, the NLM cohorts in the third line (3L) setting perform significantly better than their LM counterparts. Notably, this analysis classified only patients with no history of liver metastasis as NLM. In figure A, NLM subjects experienced a mOS of approximately 12 months and a mPFS of around 4 months, both roughly double those of LM subjects. Figure B, reporting placebo outcomes, shows that NLM patients on a placebo had an mOS of around 8 months and an mPFS of about 2 months. The corresponding LM subjects had an mOS of approximately 4.5 months and an mPFS of about 2 months.
Another exploratory analysis of mCRC patients treated with trifluridine/tipiracil also reported significantly higher survival in the NLM sub-population. The mOS in the NLM group was roughly double that of the LM cohort, 16.4 months vs. 7.7 months (Tabernero et al. 2020).
These findings underscore some striking points regarding Bot+Bal efficacy. Placebo-treated LM patients had the same (or technically slightly higher) mPFS as Bot+Bal-treated subjects. The mPFS in the NLM Bot/Bal trial is roughly the same as SOC NLM patients in the meta-analysis. Despite this, the mOS findings vary considerably; in the ICI trial, it is around 20 months, but the pooled SOC data suggests around 12 months. Why is there no change in PFS, but a huge change in OS? We argue it is due to a combination of reasons already mentioned. A small trial is likely to show exaggerated numbers due to chance, and in the Bot+Bal trial, there is significant censoring, likely informative, with patients at baseline likely being younger and possibly having a longer survival expectancy. Additionally, the LM vs. NLM mPFS data reported in the trial is comparable to data obtained from other ICIs administered in a similar setting (4.1 months vs. 4.3 months) (Fakih et al. 2020).
This highlights why interstudy comparisons are flawed, but until a proper phase 3 trial is conducted, they remain the best researchers can resort to. Agenus themselves engage in such comparisons, as seen in their 2024 press release, where they juxtapose their mCRC data with a 2023 study on trifluridine-tipiracil combined with Bevacizumab, which reported an mOS of around 13 months. This is a flawed comparison, since the 2023 trial included both LM and NLM patients, making it highly probable that the NLM subgroup would have an mOS closer to 20 months than 13 months.
Non-liver metastatic subjects are inherently “healthier” than LM patients, which may explain the observed efficacy due to confounding by indication. The hypothesis that NLM patients have more immunotherapy-responsive tumors is weakly supported, and we are skeptical of this rationale, considering the lack of proven causality and the potential for confounding factors (Tumeh et al. 2017). Furthermore, despite using the trifluridine-tipiracil + Bevacizumab trial as a comparator, the sponsor reports that the phase 2 trial, from which they intend to present data at the FDA meeting, included a standard of care (SOC) arm with either trifluridine-tipiracil or Regorafenib.
Overall, the phase 1b data presented in the journal is ambiguous, and numerous inconsistencies cast doubt over the validity of the results. We contacted the authors regarding our concerns, but have yet to receive any responses. One certainty is the regimen’s high toxicity, with 55% of patients interrupting treatment due to adverse effects. The primary regulatory issue under consideration is the toxicity associated with immune therapy, which affects many patients and significantly impacts the risk-benefit profile of the treatment. While quality of life was not assessed, it can be assumed to be poor given the high rate of adverse effects (we found one study that supposedly assessed QoL in mCRC combination ICI regimen, but did not publish the data) (Chen et al. 2020).
The censoring, especially prior to the 6-month mark, raises concerns about the validity of the data, likely violating the principle of non-informative censoring. We are skeptical about attributing the excessively high mOS to the efficacy of ICI therapy; it is more likely due to the selection of a healthier cohort. The observed responses should be verified by an external party to increase reliability. However, a ~20% overall response rate (ORR) could be attributed to chance and a small cohort of microsatellite stable (MSS) patients who do respond to ICI therapy.
The phase 2 trial that Agenus presented to the FDA was designed to enroll approximately 250 subjects and assign them to five different treatment regimens: Bot high dose, Bot low dose, Bot+Bal high dose, Bot+Bal low dose, and SOC (either trifluridine-tipiracil or Regorafenib). As mentioned earlier, the SOC offered in the trial is likely inferior to the combination therapy of trifluridine-tipiracil + Bevacizumab. Moreover, the stratification into five different cohorts negatively impacts the statistical power of the trial, although this is acceptable in an exploratory setting. Despite enrolling 250 people, each group will only include about 50 patients.
Agenus Corporate presentation May 2024 Phase 2 mCRC study design
Considering the data presented in the phase 1b trial, we doubt that Bot+Bal can outperform trifluridine-tipiracil or Regorafenib in a large-scale phase 3 trial. Likewise, the phase 2 trial is also unlikely to demonstrate ICI superiority on survival, which will likely be reported with longer median follow-up. However, with four drug groups, multiple comparisons and a heterogeneous population, the trial could significantly increase the rate of false-positive findings.
Neoadjuvant CRC
Agenus is also testing neoadjuvant Bot+Bal in CRC patients in the NEST-1 and NEST-2 trials. The controversial aspect of administering a burdensome treatment to early-stage patients prior to resection is that they are exposed to toxicity, although many will not experience recurrence regardless. For neoadjuvant therapy to be justified, there must be a significant difference in disease-free survival (DFS) or overall response (OR) shown in a large-scale trial compared to the lack of such treatment. Agenus’ early trials treated a modest number of 23 patients, who achieved notable responses to the therapy. However, similar responses have been observed with other ICIs in comparable settings (Verschoor et al. 2022).
Moreover, while the sponsor claims that no delay in time to surgery was observed, we do not believe that finding would be replicated in a large-scale trial. Even in the small cohort of the NEST-1 and NEST-2 trials, Agenus notes that two patients experienced grade 3 diarrhea/colitis, which had to be managed with immunosuppressive agents. Such occurrences are likely to be prominent in patients treated with a combination PD-1/CTLA-4 regimen, as shown in the phase 1b trial and many other trials of other ICIs, and it seems extremely unlikely that they will not impact surgical schedules. The neoadjuvant setting is still in the early stages and may seem highly promising, but we believe it is unlikely to replicate the findings in large-scale trials and is equally unlikely to yield a favorable risk-reward therapeutic profile for widespread clinical use.
mCRC BLA
We speculated that, it might be challenging to convince the FDA to advise for accelerated approval of Bot+Bal in mCRC based on the phase 1b and phase 2 trials, and not to our surprise, FDA advised against BLA filling. Not only was the phase 2 trial designed with an inferior SOC compared to the current standard, but it was also designed with low power despite enrolling approximately 250 subjects. The lack of comparisons to NLM patients in different treatment regimens creates an illusion of superior efficacy, bolstered by the small sample sizes. Interstudy comparisons and design flaws aside, the primary issue the FDA must consider is the toxicity. All approvals are based on the benefit-risk profile of the therapeutic agent. Arguably, the bar is much lower for late-stage metastatic cancer settings, where there are limited treatment options. Nonetheless, careful consideration of the outcomes observed in the phase 1b trial makes it difficult to justify early approval, an opinion with which the FDA seemingly agrees.
To fully understand the value of administering Bot+Bal to mCRC MSS patients, one can consider the results when given to 100 people. According to the phase 1b trial, 21 of those 100 people will experience a partial response, and 1 person will experience a complete response. Whether these findings translate into prolonged survival remains to be seen, as surrogate predictors of survival in cancer research are only moderately correlated. Out of the 100 people, 55 will have to interrupt the treatment due to toxicity and manage symptoms with other, likely immunosuppressive, medications. Five patients will experience grade 3 diarrhea, characterized by seven or more loose stools per day. Three people will develop grade 3 colitis, four will have grade 3 enterocolitis, and three will have grade 3 blood creatine phosphokinase increase. Many of these side effects are associated with fatality (André Thewis et al. 2024). In total, 32 patients will experience a grade 3 or higher treatment-related adverse events. This is 50% more than the number of people who will experience an OR, which, as a surrogate, is not guaranteed to predict any increase in survival. Importantly, the safety figures are reported in a cohort where one-third of patients had a median follow-up of approximately 3 months. It is reasonable to assume that with longer follow-ups, the figures will become even more pronounced.
Pancreatic Cancer
Pancreatic cancer has a reputation for being one of the most unresponsive tumor types, with poor prognostic data and short survival times (Park et al. 2021). Studies show that only about 2% of patients with this cancer type exhibit dMMR or H-MSI features, and even those patients have historically shown limited response to PD-1 therapy (approximately 17% ORR) (Timmer et al. 2021). Eli Lilly tested their PD-1 in a small cohort of pancreatic cancer patients (n=24) in combination with chemotherapy (mFOLFIRINOX) and found no difference in PFS or OS compared to chemotherapy alone, despite a significant increase in ORR (Linh Chi Tran et al. 2023). The immunosuppressive qualities of pancreatic cancer have been largely attributed to the prevalence of various markers and cell types that induce immunosuppression, including MDSCs, TAMs, CAFs, and T-regs.
Agenus is exploring Bot combined with Gemcitabine/Abraxane in patients refractory to the FOLFINOX regimen (NCT05630183). To date, data shared by the sponsor reports a total of six patients: one awaiting their first scan, one with a confirmed PR, one with an unconfirmed PR, two with SD, and one with PD who is off the study. These findings, based on early time points (8/16 weeks), show modest activity. This is an early-stage exploratory program, but there is not much to be excited about for now. We do not see Bot significantly improving outcomes in pancreatic cancer due to the historic unresponsiveness to ICI therapy, which is unlikely to be reversed by the T-reg depleting ability of Bot or increased ADCC. Furthermore, a similar regimen of Gem+Ipi was evaluated in 1L/2L pancreatic cancer patients and reported an ORR of 18%, mPFS of 2.8 months, and mOS of 6.9 months, data generally similar to 2L patient outcomes on Gem alone (Aline et al. 2015). Overall, we remain highly skeptical of the clinical success of Bot in pancreatic cancer.
Melanoma
In contrast to CRC and pancreatic cancer, melanomas are among the most responsive cancers to immunotherapy (Knight et al. 2023). Despite being an aggressive form of cancer, due to the ease of surgical excision and tumor responsiveness to therapeutic agents, melanomas have an almost 100% 5-year survival rate (Melanoma Survival Rates | Melanoma Survival Statistics 2024). To differentiate from other ICIs, Agenus chose to administer the Bot+Bal regimen to patients with melanoma that is refractory to prior PD-1/CTLA-4 therapy.
In the C-800-01 study, melanoma patients (n=10) experienced an ORR of 30%, with no complete responses observed. While this finding may seem high considering the refractory ICI status, data from other ICIs suggest similar outcomes. First-line Ipi+Nivo regimens had an ORR of 58%, although 55% of patients experienced severe toxicity characterized by grade 3+ adverse effects (Long et al. 2023). Contrary to Agenus’ findings in CRC, where NLM status indicated better responsiveness to ICIs, the study found that melanoma patients with liver involvement seemed to benefit more from the therapy. A regimen of Pembro+Ipi in patients refractory to prior PD-1/CTLA-4 therapy (n=70) reported an ORR of 29%, suggesting similar efficacy to Bot+Bal data from the solid tumors’ basket trial. Grade 3+ AEs occurred in almost a third of the patient cohort (27%) (Olson et al. 2021).
Another retrospective cohort study (n=355) reported an ORR of 31% with the Ipi+Pembro/Nivo regimen in refractory patients and 13% in patients treated with Ipi monotherapy (Pires da Silva et al. 2021). A randomized trial of patients’ refractory to PD-1 monotherapy reported an ORR of 28% for the Ipi+Nivo combo and 9% for Ipi monotherapy, largely consistent with the findings of the larger retrospective study (Vanderwalde et al. 2022). Overall, based on the available data, Bot+Bal does not appear to differentiate itself from other PD-1/CTLA-4 regimens in patients refractory to ICI therapy. The topline phase 2 trial (NCT05529316) data that Agenus intends to publish in the second half of 2024 will likely confirm these speculations.
Valuation and Cash Burn
Valuation
A biotech company is valued by the potential of its clinical assets. Accurately modeling the value of Bot and Bal is challenging, since they target a variety of cancers with different market opportunities. While we acknowledge that early-stage trials can bring value by de-risking development in a given designation, they are much less tangible than catalysts like a BLA, which will determine whether the asset will be brought to market. Therefore, in considering the NPV model of Agenus, we will focus on mCRC and Bot+Bal. This should provide an estimate of the company’s minimum value, assuming all other designations would fail. We will assess the value of mCRC Bot/Bal in NLM 3L patients using epidemiologic data, forecasting, and available data from current SOC sales in 3L patients.
Each year in the US, approximately 35 people per 100,000 are diagnosed with colorectal cancer, totaling about 100,000 individuals. Up to 50% of those people will eventually develop metastatic disease (~30% are diagnosed with metastatic disease initially and ~20% develop metachronous metastases) (Adrià Cañellas-Socias et al. 2024). About 20-30% of patients receive 3L treatment, so for our considerations, this is about 25,000 patients (Carlomagno et al. 2018) (Boyne et al. 2023). Of these, only a fraction will be classified as NLM. Literature suggests different estimates of NLM prevalence in 3L+ patients, with the company estimating 45-55%, but we believe a more conservative 25% is realistic. Hence, out of approximately 25,000 patients, about 6,000 could be eligible to receive Bot+Bal.
NPV model of Bot+Bal in mCRC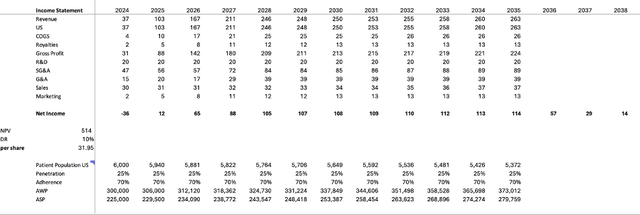
We modeled the entry of the Bot+Bal combo into the US market in late 2024, with a BLA submission anticipated in the second half of 2024, per company guidance. The sales projection includes a typical five-year ramp-up period and a 25% market penetration rate (three 3L products, market share discounted by AA uncertainty). The adherence rate was modeled at 70%, slightly below the industry average, due to toxicity and high discontinuation rates. The Average Selling Price (ASP) was modeled at 75% of the Average Wholesale Price (AWP). The initial price in the US was set at $300,000, which is the standard annual cost of PD-1/CTLA-4 therapy, with a 2% annual price increase consistent with inflation and typical for the industry.
The percent values were primarily adapted from research published in David et al. 2017. Cost of Goods Sold (COGS) was modeled at 10% of revenue, while R&D expenses were kept constant at $20 million per year to cover developmental costs associated with ongoing drug research post-market entry. Selling, General, and Administrative (SG&A) expenses were set at 34% of revenue. With a peak revenue of approximately $250 million and assuming a discount rate of 10%, the Net Present Value (NPV) of Bot/Bal in mCRC 3L was estimated to be approximately $500 million, corresponding to about $30 per share, based on a share count of 20 million, a figure which includes cash. This implies that, according to our model, the market was pricing in slightly less than a 50% probability of success (POS) for Bot+Bal AA. Note, the royalty fees payable to Ligand were not considered in these estimates.
To contextualize and verify the validity of sales estimates, we compared the modeled values with historic sales data of two drugs approved for use in 3L mCRC, Regorafenib and Lonsurf.
Regorafenib
Regorafenib is a multi-kinase inhibitor developed by Bayer, approved for 3L mCRC use in 2012. In its launch year, the drug generated $32 million in revenue, despite being marketed only in the last quarter. The following year saw approximately $200 million in Regorafenib sales, demonstrating a rapid sales launch curve, presumably partially due to being the sole treatment approved in the 3L regimen. Importantly, in 2013, the drug was also approved for 3L GIST, which might account for a significant fraction of the observed sales. Sales surpassed $300 million within five years from the drug launch but experienced a slight decrease in 2016 when another 3L treatment, Lonsurf, entered the mCRC market. Despite that, sales continued to grow, likely due to label expansion in 2017 for 2L HCC and the launch in China in 2018. Sales peaked in 2022 at $613 million, followed by a decrease of 15% in 2023, likely due to FDA approval of Lonsurf combined with Bevacizumab (data accessed from companies’ press releases and fillings).
Historic Sales of Regorafenib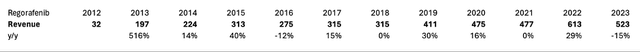
It is hard to gauge what percentage of the gross sales of Regorafenib are attributable solely to mCRC but considering $200 million sold in 2013 and approximately $300 million in 2015, prior to Lonsurf entering the market, it seems fair to estimate that Regorafenib sold around $150 million to $200 million before the HCC expansion and entering the Chinese market. Notably, Regorafenib is priced in the US at approximately $24,000 per month, amounting to almost $300,000 annually, a figure comparable to our estimated costs for Bot+Bal.
Lonsurf
Lonsurf, the brand name for trifluridine-tipiracil, is a late-stage cytotoxic drug owned by Taiho Oncology. The drug was approved for 3L mCRC in 2015 and sold approximately $60 million in its launch year. Similar to Regorafenib, it quickly ramped up to around $200 million in total sales and seemed to plateau at that level until the label expansion in 2019 for gastric cancer. Between 2019 and 2022, the sales of Lonsurf grew to approximately $350 million, and upon approval in combination with Bevacizumab for 3L mCRC, sales increased to $500 million in 2023 (data accessed from companies’ press releases and fillings). Yellow boxes represent values averaged between 2017 and 2019, due to lack of readily available data.
Historic Sales of Lonsurf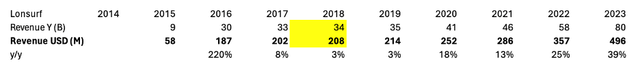
Considering the plateau at approximately $200 million prior to label expansion, in line with Regorafenib estimates, it seems likely that the peak sales of trifluridine-tipiracil in the late 2010s were around $200 million for both drugs, suggesting a total market of roughly $400 million. In the present, this figure is likely closer to $500 million, considering inflation and related price hikes.
Notably, the sales of both drugs were not restricted to NLM patients, who, as discussed previously, make up slightly less than half of the addressable population. Hence, assuming Bot+Bal could sell comparably to Regorafenib and Lonsurf in 3L mCRC, their revenue should be approximately half of these two compounds. Thus, our estimates of peak revenue of approximately $250 million might be overestimated.
Cash Burn
Historically, Agenus has been losing money, and its reported revenue has been based on licensing and milestone payments. At the end of Q1, the company had ~$50M cash, which combined with the recent licensing deal, should equal ~$125M-$150M. Their extensive pipeline and several early-stage clinical trials translate into high R&D expenses. Between 2018 and 2023, the sum of fiscal year (FY) R&D and cost of service revenue were $123M, $168M, $144M, $182M, $197M, and $237M, respectively. The G&A expenses were also relatively high between 2018 and 2023, considering the modest size of the company, standing at $37M, $46M, $59M, $76M, $81M, and $78M. While CEO Armen Garo reportedly introduced measures to drive down the company operating costs by approximately 25%, we do not believe that the licensing capital received from Ligand is sufficient to fund Agenus beyond 2024, especially considering the ongoing developments in many designations and the planned phase 3 trial of Bot+Bal in mCRC, which by itself will likely cost well over $100M.
Despite the weak stock performance, the general and administrative expenses have been consistently high. The board of Agenus is made up of six people, the majority of whom represent Agenus or related parties (Agenus – Garo Armen Ph.D; Mink Therapeutics – Jennifer Buell Ph.D, Brian Corvese, Ulf Wiinberg). Hence, it is no surprise that despite the weak performance of the stock, the board granted the CEO a bonus of over $600k in 2021, 2022, and 2023. While Dr. Armen is accepting his compensation solely in stock and stock options, his pay stood at an astounding $5.6M in 2020, almost $10M in 2021, and $5.6M and $5.8M in 2022 and 2023, respectively. In addition to the reported compensation, Dr. Armen also serves on the board of directors and owns 21% of a third-party service provider, Protagenic Therapeutics, which billed Agenus $150k, $106k, and $291k in 2023, 2022, and 2021, respectively. It is also worth noting that Dr. Armen’s son is employed at Agenus, serving as the head of investor relations.
Considering the total R&D and G&A expenses of approximately $60M in the first quarter of 2024, even if costs were significantly reduced, we still expect FY 2024 expenses to near $200M. We anticipate that the company will continue to sell off its assets through licensing or, in the worst-case scenario, resort to shareholder dilution.
Risks
There are several risks associated with our thesis, which are worth acknowledging. Most importantly, we could be wrong about the efficacy of the Bot+Bal combination. Perhaps the mechanism and the “Fc enhancement” can elicit enough activity to treat patients with cold tumors. If this turns out to be the case, our thesis could be invalid, and the drug could succeed, bringing long-term value to the company. This would not only enhance the credibility of Agenus’ pipeline but also position them as a significant player in the immuno-oncology space.
Additionally, the company could secure additional funding from milestone payments, delaying or nullifying the need for further capital raising and allowing for the execution of key developments. Given Agenus’ history of licensing deals and milestone payments, this is a plausible scenario. Successful milestones in ongoing collaborations with major pharmaceutical companies like Merck, Gilead, and BMS could provide the necessary financial support to sustain and advance their pipeline without further diluting shareholder value. Moreover, achieving significant milestones could enhance the company’s negotiating power for future deals, potentially securing more favorable terms.
Another potential risk lies in the neoadjuvant setting for CRC. While we are skeptical about its replication in large-scale trials, an unexpectedly positive outcome could bolster Agenus’ standing and open new avenues for their pipeline. Positive results in neoadjuvant settings could also drive interest in early-stage cancer treatments, further diversifying Agenus’ clinical applications.
FDA meeting and Phase 2 mCRC topline data
As noted at the beginning of this article, it was drafted prior to the release of the topline data on July 18th, 2024. In light of the new data, we decided to add an update section.
As we speculated, the FDA shared our view and discouraged the sponsor from filing a BLA through the AA pathway. This decision was based on the unlikelihood that a small ORR would translate into a clinical benefit on the OS endpoint. The ORR data released by Agenus is not highly informative, and there are several inconsistencies we would like to address.
Topline phase 2 data of Bot+Bal in mCRC – Agenus press release July 2024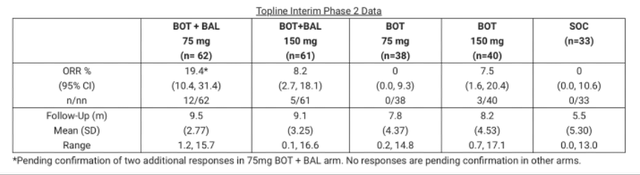
Firstly, there is a considerable imbalance in the size of the experimental arms. Even though one would expect a 1:1:1:1:1 randomization, the Bot+Bal arms enrolled approximately 60 patients, while the SOC and Bot monotherapy arms enrolled only around 30-40 patients. Furthermore, the SOC arm patients have significantly shorter mean follow-up, about half of what the ICI arms have. Most notably, however, the Bot/Bal 75 mg arm had an ORR double that of the Bot/Bal 150 mg arm. Additionally, the Bot monotherapy arm had an ORR similar to the Bot/Bal 150 mg cohort. Quite puzzling findings.
The Bot/Bal 75 mg cohort had the highest ORR, but the Bot monotherapy with the same dosing had no response. At the same time, Bot monotherapy with the high dose of 150 mg had approximately the same ORR as the Bot/Bal high dose (7.5% vs. 8.2%), as if the addition of Bal had no effect. How is it that in the high Bot dose groups, the addition of Bal seemingly had no impact on ORR, but in the low dose Bot, it increased ORR from 0% to 20%? In our opinion, there clearly is a degree of confounding or heterogeneity in the study population, which cannot be fully explained without complete data on patients and the treatments they received.
We do not interpret this as the Bot 75 mg dosing regimen being the most efficacious. We believe this inconsistency indicates a lack of profound efficacy and the low response rate typical of MSS patients treated with ICI. This data further solidifies our view on the phase 3 trial: we do not believe the Bot/Bal treatment will prove to be better than SOC in OS or PFS, which were not reported in detail in the PR. The sponsor only mentioned a “6-month survival rate for the BOT 75mg/BAL combination,” which we deem unsurprising for the NLM cohort. Furthermore, the same data point was not reported for any other arm.
Lastly, one can interpret the data comparatively to the phase 1b results published in nature medicine. The cohort in the phase 1b trial was treated with either 1 mg/kg Bot or 2 mg/kg Bot, which are ~ equal to 75mg and 150mg doses from the phase 2 trial. If one were to average ORR from the phase two, it would come down to 13.8%, less than the ORR from the phase 1b of 22% in the same NLM cohort. The shrinking ORR in our view is the result of regression to mean, and is not a promising sign of future response rate in a large-scale phase 3.
As data matures, a clearer picture of the efficacy will likely emerge. Nonetheless, as of right now, the FDA requires that Agenus carries out a large phase 3 trial to show that Bot+Bal can improve overall survival in late-stage mCRC patients.
Conclusion
To conclude, we do not believe that Agenus’ assets, namely Botensilimab and Balstilimab, will create long-term value for investors. In our opinion, their design and rationale for use in cancer patient populations non-responsive to ICI therapy is weak and will likely yield disappointment.
The company has frequently indulged in numerous early-stage expensive trials and, to finance them, has licensed the majority of its pipeline for royalties and milestone payments. The recent funding received from Ligand is unlikely to last until the end of 2024; hence, we believe that the company will have to seek additional ways to raise capital.
We do not consider the management to be reliable, given the stock performance and numerous red flags, including significant compensation despite poor performance and a questionable past.
The press release data on phase 2 Bot+Bal in mCRC is inconsistent and not highly convincing. Additionally, the FDA appears skeptical of the alleged efficacy and has discouraged the sponsor from filing for a BLA. The toxic profile of the treatment and the questionable patient stratification further undermine the credibility of the reported outcomes.
Overall, we believe that Agenus is a sell.
Read the full article here